Por Bruno Rodrigues. Doutorando do Programa de Pós-Graduação em Ciências Morfológicas, Universidade Federal do Rio de Janeiro (UFRJ) e Tecnólogo do Instituto de Biodiversidade e Sustentabilidade NUPEM – UFRJ-Macaé
A manutenção da vida em suas diferentes formas é per si um imenso desafio quando levada em consideração as diferentes condições ambientais existentes em nosso planeta. Diferentes espécies se adaptaram com o passar dos anos a diferentes condições (e continuam se adaptando nos dias de hoje) e, caso tal adaptação não seja feita de uma forma eficaz, as consequências podem ser extremas ao ponto de sua extinção.
Na busca de uma melhor compreensão de como organismos podem regular seu metabolismo ao serem submetidos a estresses ambientais, um grupo interdisciplinar liderado pelo pesquisador Joshua Rosenthal estudou como uma espécie de polvo consegue se adaptar a grandes variações de temperatura da água na qual eles vivem.
A espécie Octopus bimaculoides é um tipo de polvo poiquilotérmico, isto é, que possui grande variação em sua temperatura corporal. A dificuldade em uma rápida resposta variação de temperatura poderia ter severos impactos em sua fisiologia. Neste sentido, os pesquisadores se perguntaram o quanto edições no RNA mensageiro (RNA-m), uma importante molécula intermediária na propagação da informação contida no DNA dos seres vivos, poderia ser importante para o sistema nervoso do animal em estudo, tendo em vista que estas edições são muito mais prevalentes nestes animais.
Para responder esta pergunta, os pesquisadores focaram em edições em uma específica molécula contida no RNA, as adenosinas. Esta edição é interessante pois seu produto se torna interpretável como uma diferente molécula também presente no RNA – a guanosina – podendo ao final ter implicações no resultado gerado por estas.
Com isto, os pesquisadores submeteram os animais a diferentes condições de temperatura, enquanto um grupo de polvos foi aclimatado a uma temperatura de 22C, um outro grupo foi submetido a uma temperatura de 13C pelo mesmo período. Após o período de exposição a diferentes temperaturas, os pesquisadores coletaram uma parte específica do seu sistema nervoso previamente conhecida por possuir altas taxas destas edições chamada gânglio estrelado, e desta eles recolheram todas as suas moléculas de RNAm existentes para que então pudessem estimar quantas adenosinas passariam a ser interpretadas como guanosinas.
Para a surpresa do grupo de pesquisadores, aproximadamente 33% das edições encontradas nas moléculas de RNAm eram oriundas dos animais submetidos a baixa temperatura, enquanto apenas 1% destas sequências foram encontradas nos animais submetidos a temperaturas mais elevadas.
Mas como estas edições ajudariam na regulação fisiológica deste polvo quando desafiados a temperaturas mais frias? Em busca desta resposta, os pesquisadores selecionaram duas importantes proteínas contidas neste tecido e que tiveram sua estrutura alterada por conta destas edições, e realizaram ensaios para avaliar o impacto destas alterações em suas respectivas funções. Como resultados eles observaram que, de fato, a alteração da estrutura destas proteínas influenciam suas funções na neurofisiologia do organismo, e que estas alterações podem ser observadas tanto em laboratório quanto nos animais que estão em seu habitat natural.
Com isto, este estudo ajuda na elucidação de diferentes estratégias adotadas por organismos na busca de adaptações ao ambiente no qual estão expostos de uma forma até então pouco explorada, além de despertar o imaginário dos estudiosos da vida sobre suas implicações em diferentes estímulos e do por que estas edições serem mais comuns nestes fascinantes animais que são os polvos.

Figura: Gráfico resumindo os principais achados do estudo de Birk et al., 2023 Cell. Os eventos de recodificação induzidos pela mudança de temperatura e seus efeitos que levam a alterações nas funções de proteínas.
Leia mais no artigo original em:
Temperature-dependent RNA editing in octopus extensively recodes the neural proteome
Matthew A. Birk, Noa Liscovitch-Brauer, Matthew J. Dominguez, R. Bryan Sutton, Eli Eisenberg
Joshua J.C. Rosenthal. Cell VOLUME 186, ISSUE 12, P2544-2555.E13, JUNE 08, 2023
Trabalho de pesquisadores do instituto de Genômica Funcional de Lyon demonstra a complexidade da análise comparativa do processo de desenvolvimento e regeneração num mesmo organismo.
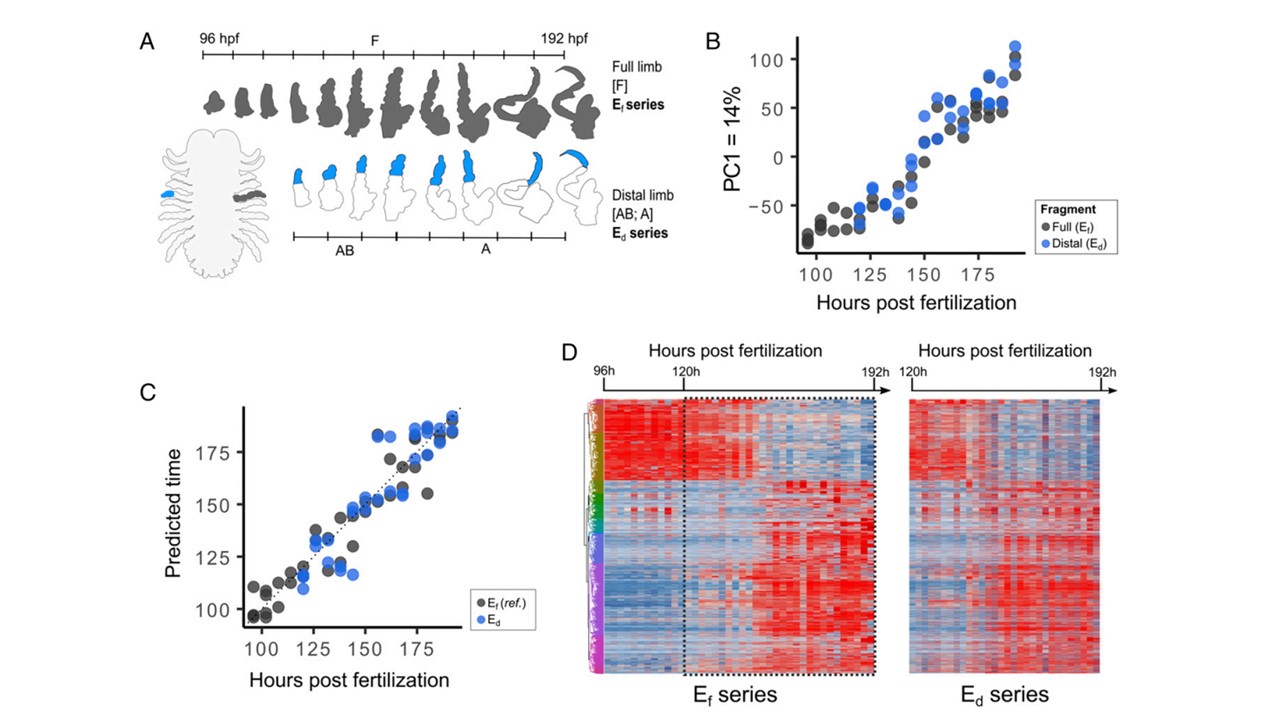
Picture from Distinct gene expression dynamics in developing and regenerating crustacean limbs Chiara Sinigaglia, Alba Almazan, Marie Lebel, Marie Semon, Benjamin Gilleta, Sandrine Hughesa, Eric Edsingerc, Michalis Averof, and Mathilde Paris. PNAS 2022 Vol. 119 No. 27 e2119297119
Nem todos os animais conseguem regenerar suas estruturas após um processo de injúria como por exemplo a perda de uma perna causada pela ação de um predador, ou mesmo numa luta entre indivíduos. Nós humanos somos um exemplo disso, após a perda de um membro num acidente nossos tecidos não conseguem se reorganizar e regenerar o membro perdido mesmo com todo conhecimento da medicina regenerativa atual.
Ao contrário dos humanos, o crustáceo Paryhale hawaensis consegue restabelecer uma perna completa após um processo de amputação em cerca de cinco dias, um tempo bastante parecido com o seu processo natural de desenvolvimento de uma perna.
Assim, o grupo de Michalis Averof e Matilde Paris questionou se os processos de desenvolvimento e de regeneração das pernas seguiriam uma lógica molecular similar, isto é, se os mesmos grupos de genes seriam expressosa numa ordem cronológica similar entre os grupos de pernas em desenvolvimento e em regeneração. Para isto, o grupo realizou análises de transcriptoma comparativab com pernas em desenvolvimento e em regeneração, gerando conclusões bastante interessantes sobre a comparação dos dois processos. Interessantemente, os transcriptomas das pernas em desenvolvimento de diferentes indivíduos são muito parecidos entre si, enquanto os transcriptomas de pernas em processos de regeneração variam bastante entre os indivíduos. Os autores buscaram tentar entender essas diferenças nas pernas em regeneração e observaram que o estágio do ciclo da muda do animal de cerca de 27 dias é responsável por uma grande mudança na regulação transcricional. Isto é, indivíduos amputados no quinto dia após a muda tem um perfil de transcriptoma parecido entre si, assim como indivíduos próximos a muda (>20 dias após a última muda), logo os sinais de expressão regenerativos acabam sendo pouco evidentes nesses animais devido as diferenças no tempo do ciclo de muda dos animais.
Assim, o estudo é extremamente interessante pois demonstra que a hipótese de que o processo de regeneração seria simplesmente uma desdiferenciação do tecido que permanece no local da lesão seguida dos processos normais de desenvolvimento embrionários como proliferação, padronização e diferenciação provavelmente não é uma conclusão tão simples e direta. Mais estudos são necessários para identificação de mecanismos comuns entre os processos de desenvolvimento e regeneração, sendo a regeneração da perna deste crustáceo um excelente modelo para esta comparação.
a – Utiliza-se o termo o “gene ser expresso” para caracterizar o processo de transcrição de uma sequência de DNA para RNA. Este processo ocorre em todas a as células ao longo do desenvolvimento e da regeneração sendo o controle da expressão gênica um dos principais eventos responsáveis por mudanças nos estados de proliferação e diferenciação celular.
b – O transcriptoma é o conjunto global de sequencias de RNA que são transcritas (de DNA para RNA) a partir de um determinado tecido ou célula. Com as metodologias de sequenciamento de nova geração é possível obter milhões de sequencias transcritas a partir de um único tecido ou mesmo única célula fazendo com que possamos comparar a expressão de um grande conjunto de genes (>10000) de uma só vez.
Leia mais no artigo original em:
Distinct gene expression dynamics in developing and regenerating crustacean limbs Chiara Sinigaglia, Alba Almazan, Marie Lebel, Marie Semon, Benjamin Gilleta, Sandrine Hughesa, Eric Edsingerc, Michalis Averof, and Mathilde Paris. PNAS 2022 Vol. 119 No. 27 e2119297119. https://doi.org/10.1073/pnas.2119297119
Texto da aluna Poliana Landry de Almeida Silva (Aluna de Iniciação Científica do Instituto NUPEM/UFRJ-Macaé)
O artigo “Evidence of multifaceted functions of codon usage in translation within the model beetle Tribolium castaneum”, publicado na revista DNA Research, aborda sobre o uso de códons em genes altamente transcritos na linhagem germinativa (ovário e testículos) e nos tecidos somáticos (machos e fêmeas estéreis) do besouro Tribolium castaneum. A transcrição é o primeiro estágio da expressão do gene no qual será produzida a fita de RNA mensageiro (RNAm). Os códons presentes nessa fita são grupos de três nucleotídeos, sendo estes compostos pelas bases nitrogenadas: Adenina, Citosina, Uracila e Guanina. A leitura do RNAm, processo denominado como tradução, irá decodificar uma proteína a partir da codificação de cada códon que determinará um aminoácido ou indicará o ponto de início ou fim da tradução. Os RNAs transportadores (tRNAs) são os responsáveis em fazer a decodificação de um códon em um aminoácido específico a partir da ligação entre anticódon (extremidade inferior do tRNA) e códons específicos do RNAm. Na prática, o reconhecimento de um códon por um tRNA pode sofrer o fenômeno de oscilação. Um pareamento de bases não padrão pode se formar entre o terceiro nucleotídeo do códon e o primeiro nucleotídeo do anticódon. Esse fenômeno permite que um tRNA leia dois ou possivelmente três códons. Em geral, o resultado do genoma será o conjunto de proteínas funcionais.
Com base em estudos realizados com aranhas, insetos e bactérias, os genes altamente transcritos, geralmente, utilizam um subconjunto de códons chamados de códons ideais que são os códons mais usados nos genes com alta transcrição. Mas, também, existem os códons não ideais (códons menos usados em genes altamente transcritos) que podem desempenhar papéis significativos no processo de tradução. Dessa forma, o artigo mostra que a vasta maioria dos códons ideais primários eram os mesmos em cada um dos tipos de tecidos. Porém, foi observado uma minoria de códons ideais variando nos quatro tipos de tecidos, sugerindo diferenças pequenas, mas potencialmente significativas. Por exemplo, um códon ideal primário específico do sexo masculino foi identificado para o aminoácido Fenilalanina, pois o códon TTC foi ideal nos testículos e machos estéreis, mas não nos ovários ou fêmeas estéreis. Os resultados também sugerem uma tendência de todo o genoma para maior uso de códons ideais em regiões codificadoras com maiores expressões.
O artigo expõe duas hipóteses sobre os códons. A primeira mostra que, embora, os códons ideais identificados sejam preferidos em genes altamente transcritos, seu benefício inato não pode ser devido ao fato de terem altas quantidades de tRNAs de correspondência direta e que, portanto, deve-se empregar o tRNA de oscilação. Os códons que utilizam tRNA de oscilação podem desempenhar papéis supostamente significativos na tradução do mRNA. Além disso, certos códons não ideais, neste táxon, não estão inevitavelmente ligados a uma baixa abundância de genes de tRNA correspondentes e, em vez disso, exibiram altas contagens de genes de tRNA correspondentes. A segunda hipótese propõe que o uso de códons não ideais com várias cópias de genes de tRNA é um mecanismo que promove alta tradução de mRNA de genes com funções celulares específicas. Assim, o estudo revela a complexa dinâmica do uso de códons no besouro multicelular T. castaneum.

Artigo original disponível em:
Carrie A Whittle, Arpita Kulkarni, Cassandra G Extavour. Evidence of multifaceted functions of codon usage in translation within the model beetle Tribolium castaneum. DNA Research, 2020.
Por Mateus Berni, Laboratório de Biologia Molecular do Desenvolvimento (Helena Araujo’s Lab), Programa de Ciências Morfológicas (PCM, Instituto de Ciências Biomédicas), Universidade Federal do Rio de Janeiro
O genoma da rã africana Xenopus laevis, um dos anfíbios mais comuns em laboratórios de pesquisa e organismo utilizado em importantes descobertas científicas na área de biologia celular e biologia do desenvolvimento acaba de ter seu genoma sequenciado. O estudo liderado por cientistas da Universidade da Califórnia Berkeley (EUA) e da Universidade de Tóquio (Japão), contou com a participação de cientistas de inúmeros outros países e foi matéria de capa da revista “Nature” (20 Outubro 2016)
O estudo lança luz em relação à origem da espécie e da poliploidia (possuir mais do que dois pares de cromossomos homólogos) de Xenopus, sendo essa descoberta incomum entre os vertebrados. Dados moleculares sugerem que uma espécie ancestral diplóide de Xenopus se divergiu de outra há aproximadamente 34 milhões de anos. Essas duas populações ancestrais de Xenopus, se tornaram duas espécies distintas, denominadas figurativamente aqui de espécie A e espécie B, e evoluíram independentemente por 17 milhões de anos. Não se sabe ao certo o porquê essas duas espécies se intercruzaram, mas deste intercruzamento originou-se Xenopus laevis, uma espécie tetraplóide, ou seja, com quatro cópias de cada cromossomo, duas vindas de cada espécie (veja a extended figure 1a do artigo original para as possíveis causas da poliploidização).
Como os cientistas chegaram a esta conclusão? Uma das evidências se baseia em sequências parasitas chamadas elementos transponíveis, que são sequências que se inserem randomicamente no DNA. Durante o tempo em que as espécies A e B evoluíram independentemente, os cromossomos destas espécies foram parasitados por diferentes famílias de elementos transponíveis. E mesmo após a fusão das espécies, os diferentes cromossomos das espécies A e B não se misturaram. Desta forma, durante o sequenciamento do genoma, algumas famílias de elementos transponíveis estavam presentes somente em um grupo de cromossomos chamado L, associado à espécie A, e outras famílias de elementos transponíveis infectaram apenas o grupo denominado S, associado à espécie B. É isto mesmo, mesmo tendo passado aproximadamente 17 milhões de anos da fusão entre as espécies, ainda é possível identificar a origem de cada conjunto de cromossomos.
Agora que o novo organismo possui 2 conjuntos de cromossomos, isto é, cerca de duas vezes mais genes que as espécies ancestrais, o que acontece com o “excesso” de genes em um organismo poliplóide? Uma série de artigos afirmam que genes com a mesma função em um organismo poliplóide são rapidamente covertidos à cópias simples, através de mutações ou deleções em um dos genes duplicados, exceto quando adquirem novas funções. Desta forma, apenas deixando o tempo atuar sobre um organismo poliplóide, ocorreria um processo de diploidização, isto é, a perda da maioria dos elementos redundantes. Em Xenopus, por outro lado, mais de 56% dos genes foram mantidos em ambos os grupos cromossômicos (L e S). Esta incrível retenção de genes duplicados fez com que fossem encontrados mais de 45 mil genes no genoma de X. laevis, ante cerca de 25 mil genes no genoma humano.
Um dos pontos mais interessantes do artigo mostra que estes grupos de cromossomos estão evoluindo assimetricamente em relação ao outro. Quando o genoma de X. laevis é comparado com uma espécie diplóide próxima chamada Xenopus tropicalis, nota-se que enquanto o grupo cromossômico L perdeu cerca de 8,5% dos genes, o grupo S perdeu mais de 31,5 % dos genes, quase quatro vezes mais deleções. Fato diferente do que ocorreu com o a truta arco-íris, outro organismo tetraplóide que teve seu genoma sequenciado, e que apresentou uma perda gênica uniforme entre entre seus cromossomos. Além disto, quando se analisa a sintenia dos cromossomos, isto é, a ordem dos genes entre os três conjuntos gênicos: Xenopus tropicalis, X. laevis conjunto L e X. laevis conjunto S, é encontrada uma grande similaridade entre as posições dos genes do cromossomo de X. tropicalis com o conjunto L de X. laevis, já o conjunto S de X. laevis além de apresentar um maior número de deleções, apresenta uma série de translocações, isto é, inversões em porções dos cromossomos (figura 1). Veja que interesante, mesmo após a união do genoma de duas espécies distintas em um mesmo núcleo, seus respectivos cromossomos continuam a evoluir separadamente, sendo que aparentemente o conjunto L mantém a condição ancestral e S apresenta maior número de modificações.
Outra importante descoberta do estudo, foi sobre as diferentes taxas de retenção e perda de genes em relação às suas funções. Genes envolvidos na regulação gênica, por exemplo fatores de transcrição, tiveram um alta taxa de retenção (>90%), sendo que 37 dos 38 genes do grupo homeobox foram mantidos. Por outro lado, genes envolvidos no reparo do DNA e genes relacionados ao metabolismo tiveram uma taxa de deleção mais alta. Estes dados indicam que genes onde a dose é importante, isto é, onde os níveis de expressão precisam ser finamente controlados, a taxa de retenção é mais alta, e em genes onde o fino controle da taxa de expressão não é extremamente essencial, a taxa de perda gênica é mais elevada.
O dados do genoma de X. laevis levanta uma série de hipóteses, entre elas, de que o “excesso” de DNA não é tão danoso para um organismo, e a taxa de deleção do “excesso de informação” é extremamente lenta, o que confere tempo para que essas sequências redundantes possam adquirir novas funções. O sequênciamento do genoma de outros organismos poliplóides pode fornecer inúmeras pistas sobre a evolução do genoma dos vertebrados, visto que existem fortes evidências de que neste grupo ocorreram dois processos de poliploidização no início de sua irradiação, há cerca de 500 milhões de anos.
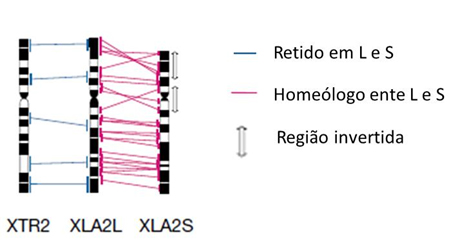
Figura 1. Arranjo sintênico do cromossomo 2 de X. tropicalis (XTR) e dos dois grupos cromossômicos de X. laevis (XLA2L e XLA2S). Note que o cromossomo de X. tropicalis apresenta grande conservação entre a ordem dos genes com o conjunto de cromossomos L de X. laevis, e já o conjunto S de X. laevis, apresenta rearranjos e deleções na organização cromossômica. Nota:homeólogo são cromossomos parcialmente homólogos, com uma ancestralidade em comum. Adaptado de Session et al 2016
Leia mais em: http://www.nature.com/nature/journal/v538/n7625/abs/nature19840.html
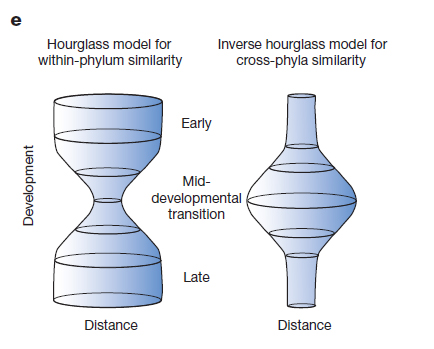
Figura 1: Conservação gênica e comparações intra-Filo e inter-Filo do desenvolvimento embrionário.
O Reino Animalia (Metazoa) é constituído por 38 grupos monofiléticos (Filos) segundo o último livro do respeitado taxonomista Claus Nielsen (Animal Evolution, Oxford). O número de Filos e suas relações filogenéticas são motivo de bastante discussão na literatura científica com dados morfológicos e moleculares sendo, muitas vezes, divergentes entre si.
Quando um professor no ensino médio entra na sala de aula e introduz os Filos como por exemplo, Porifera, Cnidaria, Placozoa dentre outros, uma questão sempre vem a mente dos alunos. O que caracteriza um Filo? Livros e publicações tradicionais definem Filos como grupos de organismos que possuem a organização corporal comum, assim, organismos que possuem organização corporal distinta devem estar agrupados em Filos diferentes.
Nos últimos anos, diversos estudos de análise de expressão global ao longo do desenvolvimento (transcriptomas) em organismos-modelo como a mosca-da-fruta (Drosophila) e peixe-zebra (“zebrafish”) demonstraram que genes mais antigos na evolução dos animais são expressos no chamado estágio filotípico, em inglês “phylotypic stage” quando comparações são feitas dentro do mesmo Filo (Domazet-Lošo, T. & Tautz, D. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468, 815–818 (2010). ). Logo, segundo este e outros estudos, os genes mais antigos na evolução dos animais, incluindo os fatores de transcrição da família HOX, seriam expressos no estágio filotípico, isto é, o estágio em que os animais de um mesmo Filo se pareceriam bastante morfologicamente.
Na edição de Março da revista Nature, Levin et al., 2016 realizaram uma abordagem diferente. Ao invés de compararem a expressão de genes (transcriptomas) de organismos de um mesmo Filo, Drosophila nos artrópodes, ou peixe-zebra nos vertebrados, Levin et al., realizaram uma comparação da expressão global dos genes (transcriptomas) entre organismos de 10 diferentes Filos dos animais incluindo Porifera, Cnidaria, Nematoda, durante a embriogênese. Ao contrário dos resultados obtidos com comparações dentro dos Filos, em comparações de expressão entre Filos foi observada grande variação dos genes expressos durante o estágio intermediário, o estágio Filotípico. Na comparação entre Filos, os genes expressos no início e no final do desenvolvimento seriam conservados (Figura 1).
O artigo com maiores informações pode ser obtido abaixo:
 A embriogênese dos animais pode ser dividida em duas estratégias conhecidas como desenvolvimento embrionário determinado e regulativo. Estas duas formas de desenvolvimento embrionário foram propostas a cerca de 100 anos atrás por dois discípulos do famoso cientista alemão Enrst Haeckel, Hans Driesch e Wilhelm Roux. Roux e Driesch foram dois dos grandes precursores da embriologia experimental atual. Os estudos desses dois pesquisadores com manipulação de embriões de sapo e ouriço-do-mar demonstraram, de formas diferentes, que ovos de animais podem ter comportamentos diferentes quando submetidos a agentes estressantes como mudanças de temperatura, agentes causadores de mutações (radiação ultravioleta-UV), ou fragmentação por processos mecânicos.
A embriogênese dos animais pode ser dividida em duas estratégias conhecidas como desenvolvimento embrionário determinado e regulativo. Estas duas formas de desenvolvimento embrionário foram propostas a cerca de 100 anos atrás por dois discípulos do famoso cientista alemão Enrst Haeckel, Hans Driesch e Wilhelm Roux. Roux e Driesch foram dois dos grandes precursores da embriologia experimental atual. Os estudos desses dois pesquisadores com manipulação de embriões de sapo e ouriço-do-mar demonstraram, de formas diferentes, que ovos de animais podem ter comportamentos diferentes quando submetidos a agentes estressantes como mudanças de temperatura, agentes causadores de mutações (radiação ultravioleta-UV), ou fragmentação por processos mecânicos.
Experimentos como estes nas décadas subsequentes demonstraram que ovos da mosca-da-fruta, um inseto, tem o desenvolvimento do tipo determinado, isto é, quando uma região destes ovos é submetida à radiação UV, esta região é perdida e o embrião não consegue “recuperar” essa parte perdida. Nem todo ovo de inseto possui desenvolvimento determinado, como mostrou o embriologista alemão Klaus Sander entre 1950-70. Sander colocou ovos do hemiptera Eucelis numa pequena gilhotina e dividiu cada ovo em metades que permaneciam conectadas pela camada envoltória, chamada de córion. Sander observou que, em várias situações, eram formados dois embriões menores, porém completos um em cada metade (Figura abaixo).
Ao contrário do desenvolvimento determinado observado em Drosophila, o ovo de hemiptera demonstrou desenvolvimento regulatório, isto é na perda de uma parte do embrião, as demais células foram capazes de se regular, gerando novas partes e assim formaram um embrião inteiro.
A grande questão seria como estes dois embriões são gerados numa situação em que teoricamente deveria haver um só ? Em recente artigo na revista eLIFE Sachs et al., 2015 demonstraram que duas vias de sinalização bem conhecidas, a via de Toll e a via de BMP (Figura) são fundamentais para explicar como esse fenômeno acontece. O que Lachs et al descobriram é que neste hemiptera, um “primo” próximo do vetor da doença de chagas Rhodnius prolixus que estudamos no laboratório, a via de sinalização de BMP é essencial para induzir a formação de todo o eixo que padroniza a parte ventral (barriga) e dorsal (costas) deste inseto, e que a via de Toll tem papel pequeno e transitório neste processo. Utilizando diversos resultados experimentais, e um modelo teórico elaborado Lachs et al., conseguiram explicar, pelo menos em teoria, como um embrião regulativo se comporta e como dois embriões são formados nas condições estabelecidas por Sander na década de 70.
Vale a leitura!
Dynamic BMP signaling polarized by Toll patterns the dorsoventral axis in a hemimetabolous insect
Lena Sachs,1,† Yen-Ta Chen,1,† Axel Drechsler,1,2 Jeremy A Lynch,1,3 Kristen A Panfilio,1 Michael Lässig,4 Johannes Berg,4 and Siegfried Roth1,*
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4423117/

De 6 a 8 de abril pesquisadores brasileiros e internacionais de Evo-Devo se reuniram no Centro de Genômica e Biologia de Sistemas da Universidade Federal do Pará, Belém para um encontro envolvendo alunos de graduação e pós-graduação do Programa de Pós-Graduação em Genética e Biologia Molecular (PPGBM-UFPA) e do Programa de Pós-Graduação em Neurociências e Biologia Celular (UFPA).
O Encontro científico de altíssima qualidade só foi possível devido a exemplar organização de Igor e Patricia Schneider (UFPA). O meeting contou com a participação de dois excelentes convidados estrangeiros no Brasil, a partir de verba do Programa Ciências Sem Fronteiras, Chris Amemiya (University of Washington, Seattle, USA) e John Taylor (University of Victoria, Canada). Estes pesquisadores internacionais com certeza sairão do Brasil com uma excelente impressão das instalações do Centro de Genômica e Biologia de Sistemas, do entusiasmo dos alunos da UFPA com a Evo-Devo, e com excelentes colaborações com pesquisadores nacionais. Esse tipo de iniciativa do Ciências sem Fronteiras deve ser mantido e expandido em particular em áreas novas e pouco difundidas no nosso país como a Evo-Devo.
A diversificada programação do evento encontra-se abaixo e fico muito feliz de ver que a qualidade das palestras dos brasileiros em nada deveu a dos estrangeiros. Podemos colaborar e gerar novos conhecimentos sem abaixar a cabeça, pois nossos trabalhos possuem nível internacional. Este meeting é mais um passo para construir uma comunidade jovem e diversificada de Evo-Devo no Brasil. Temos modelos e perguntas biológicas importantes.
E você que está lendo o post não quer fazer parte dessa comunidade de Evo-Devo no Brasil ?
Texto escrito por Ivan Dias, Laboratório de Evolução Marinha (LEM/USP)/Laboratório de Biologia Evolutiva do Desenvolvimento (EvoDevo Lab/USP), Instituto de Biociências, Universidade de São Paulo (IB-USP), aluno de Federico Brown, USP.

Estes parecem ser tempos controversos para os biólogos evolutivos. De acordo com um comentário recentemente publicado na renomada Nature, uma revista científica semanal, pesquisadores estão divididos sobre quais processos devem ser considerados fundamentais para a evolução.
A atual Teoria da Evolução foi forjada entre as últimas décadas do século 19, com os importantes trabalhos de Charles Darwin (1859–1872), e os anos 1930 e 1940, quando vários estatísticos e geneticistas combinaram o conceito darwinista de seleção natural com os campos emergentes da genética e, em menor escala, da paleontologia e da sistemática, em um consenso sobre como ocorre a evolução biológica. A chamada “síntese moderna”, ou neodarwinismo, lançou as bases teóricas que permitiram que o processo evolutivo passasse a ser descrito matematicamente, como mudanças nas frequências de variantes genéticas em uma população, ao longo do tempo.
De acordo com essa síntese moderna, novas variações surgem através de mutações genéticas aleatórias, a herança ocorre através do DNA e a seleção natural na luta pela vida torna alguma(s) dessas variações adaptativas mais comuns, por meio da sobrevivência e reprodução diferenciais. A seleção natural é a única causa de adaptação, processo através do qual as espécies se tornam bem adaptadas aos seus ambientes. Esta síntese moderna ainda reconhece a importância de outros processos como a deriva genética – mudanças aleatórias na frequência das variantes genéticas devido à amostragem – e o fluxo gênico entre as populações, responsável pela entrada ou saída de variantes de uma população, via migração, dispersão ou acasalamento.
Alguns pesquisadores, no entanto, argumentam que essa síntese é “genocêntrica” (centrada nos genes) e “não consegue captar toda a gama de processos que direcionam a evolução”. De acordo com esses biólogos, há algumas peças faltantes na atual teoria da evolução, que incluem vários processos importantes, mais recentemente descritos e evidenciados por diferentes áreas das ciências biológicas, como a biologia do desenvolvimento e a ecologia evolutiva.
Dois desses processos evolutivos importantes seriam os viéses de desenvolvimento, que significam que o desenvolvimento físico influencia a geração de variação – processos de desenvolvimento que orientam as formas dos organismos ao longo de vias particulares; e a plasticidade fenotípica, ou como o ambiente molda diretamente as características dos organismos: novas formas, potencialmente funcionais, são induzidas pelo ambiente e posteriormente estabilizadas pela seleção.
Outros processos candidatos à essa atualização, de acordo com esses pesquisadores, são a construção de nicho, ou como os organismos modificam sistematicamente os recursos ambientais de forma que impõem desvios sobre a evolução e o desenvolvimento de seus descendentes, das barragens construídas pelos castores aos solos processados por vermes – e a herança inclusiva, ou como os organismos transmitem mais do que genes através das gerações, podendo herdar uma grande variedade de materiais de seus antepassados, incluindo marcas epigenéticas (alterações químicas que alteram a expressão do DNA, mas não a seqüência de bases subjacente), hormônios, simbiontes, comportamento socialmente transmitido nos animais (conhecimentos e habilidades aprendidas) e legados ecológicos (estruturas e condições ambientais alteradas que os organismos deixam a seus descendentes através da construção de nicho). Essas heranças têm grande influência sobre a fertilidade, a longevidade e a resistência a doenças nos mais diversos táxons.
Outros biólogos evolutivos, embora reconhecendo a sua importância, afirmam que esses quatro processos não precisam de uma atenção tão especial a ponto de merecerem uma revisão da atual teoria da evolução sob o nome de uma “síntese evolutiva estendida”. De acordo com eles, esses processos não são negligenciados pelos biólogos evolutivos e já estão bem integrados à estrutura conceitual da area. Quanto à crítica de que a teoria da evolução atual é “genocêntrica”, os defensores da síntese moderna argumentam que, de fato, os genes são fundamentais, com as mudanças no material hereditário sendo uma parte essencial dos processos de adaptação e especiação. Os processos que os revisores defendem só podem participar na evolução, se houver variação genética associada a eles. Parece que o que ainda importa, afinal, são as diferenças hereditárias das variantes, especialmente as que conferem alguma vantagem seletiva – e, portanto, o argumento central da teoria da evolução de Darwin permanece.
E você, de que lado dessa controvérsia científica você está?
Laland, K. et al. 2014 Does evolutionary theory need a rethink? Nature 514, 161–164.
 Foi realizado entre 21 e 25 de julho de 2014 a quinta-edição do Encontro da Sociedade Européia de Biologia Evolutiva do Desenvolvimento (Euro Evo-Devo) um meeting vibrante e com um imenso número de modelos e perguntas que fascinam biologistas evolutivos do desenvolvimento do mundo inteiro incluindo um grupo de brasileiros. Com 600 participantes pode-se dizer que 1% do congresso foi composto por brasileiros, pois eram literalmente seis dos quais três estão representados na foto abaixo, eu Rodrigo Nunes da Fonseca do Núcleo em Ecologia e Desenvolvimento Sócio-Ambiental de Macaé (NUPEM) da Universidade Federal do Rio de Janeiro, a Profa. Tiana Kohlsdorf da Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, USP-Ribeirão Preto e a pós-doutora Juliana Roscito do Max Planck Institute of Molecular Cell Biology and Genetics (Foto).
Foi realizado entre 21 e 25 de julho de 2014 a quinta-edição do Encontro da Sociedade Européia de Biologia Evolutiva do Desenvolvimento (Euro Evo-Devo) um meeting vibrante e com um imenso número de modelos e perguntas que fascinam biologistas evolutivos do desenvolvimento do mundo inteiro incluindo um grupo de brasileiros. Com 600 participantes pode-se dizer que 1% do congresso foi composto por brasileiros, pois eram literalmente seis dos quais três estão representados na foto abaixo, eu Rodrigo Nunes da Fonseca do Núcleo em Ecologia e Desenvolvimento Sócio-Ambiental de Macaé (NUPEM) da Universidade Federal do Rio de Janeiro, a Profa. Tiana Kohlsdorf da Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, USP-Ribeirão Preto e a pós-doutora Juliana Roscito do Max Planck Institute of Molecular Cell Biology and Genetics (Foto).
Quais as principais novidades no mundo da Evo-Devo que o congresso demonstrou e que deve ser divulgada para novos estudantes e outros pesquisadores do país ? Aqui eu destaco três elementos que com certeza estão e estarão revolucionando o mundo da Evo-Devo.
O primeiro e mais abrangente é o grande número de estudos envolvendo sequenciamento de DNA em larga escala, ou seja a obtenção de muitas sequencias de DNA de um organismo, a um preço acessível. Para entender a dimensão desse fenômeno chegamos a preços acessíveis até a pesquisadores brasileiros que muitas vezes não possuem muitos recursos. A associação de estudos de transcriptoma, isto é, a obtenção de todas as sequencias de RNA transcritas em um determinado instante com estudos de perda de função de genes está revolucionando a Evo-Devo moderna.
O segundo ponto é a importante inclusão de aspectos ecologicos e fisiológicos na compreensão dos mecanismos moleculares que estão envolvidos na evolução de novas formas. Por exemplo, ao analisar espécies relacionadas numa dada filogenia, mas que possuem diferenças morfológicas importantes para a vida num determinado ambiente busca-se compreender como essas novidades evolutivas surgiram ao nivel genético.
O terceiro ponto seria a busca mais conceitual dos principais conceitos advindos dos últimos anos nos estudos de Evo-Devo onde questões conceituais como evolvabilidade, pleiotropia e restrições filogenéticas foram discutidas intensamente.
Uma última novidade importante é para os latino americanos é a criação da Sociedade Pan Americana de Biologia Evolutiva do Desenvolvimento https://www.hub.ki/groups/sedb. Os interessados podem se inscrever na sociedade no link ao lado. O secretário latino-americano é o Prof. Federico Brown (USP) e esta sociedade estará realizando seu primeiro encontro em Agosto de 2015 em Berkeley. Estaremos certamente lá.


Em um artigo clássico na década de 1940 o embriologista C.H. Waddington propôs o termo canalização para explicar o fato de um organismo ser capaz de produzir sempre as mesmas características (fenótipo) mesmo com as variações externas do meio ambiente e os possíveis ruídos. Waddington exemplificou este fenômeno de canalização usando a trajetória de uma bola navegando ao longo de uma região de montanhas e vales. Com o passar do tempo a bola acaba sendo levada (canalizada) para a região de vale e portanto os vales seriam os canais em que o desenvolvimento embrionário seria direcionado.
Em um recente artigo na respeitada revista Current Biology, o laboratório de Chip Ferguson demonstrou evidências para a canalização, a partir da análise do desenvolvimento embrionário da mosca-da-fruta Drosophila melanogaster. Esta mosca padroniza sua região dorsal a partir da sinalização de uma proteina bem importante para os animais, a BMP (Bone Morphogenetic Protein). Embora tenha o nome de padronizadora de osso esta proteina atua em diversos processos da vida da mosca e na nossa também como na padronização das células extra-embrionárias.
Ao analisar um tipo de duplo mutante que não tem reguladores fundamentais de BMP, Ferguson e seu grupo observaram uma grande variação na atividade de BMP e consequentemente no número de células extra-embrionárias das moscas. Assim, na Drosophila selvagem existe um sistema genético robusto que leva a canalização. Este sistema genético envolve pelo menos quatro moléculas com efeitos diferentes que canalizam o sistema fazendo com que o desenvolvimento da mosca seja correto e pouco variável. Waddington ficaria feliz ao ler este artigo 60 anos depois de suas descobertas !
Development: getting into the groove, or evolving off the rails? – Comentário sobre o artigo
Panfilio KA, Roth S. Curr Biol. 2013 Dec 16;23(24):R1101-3. doi: 10.1016/j.cub.2013.10.073.
PMID: 24355787 [PubMed – in process]
A genetic network conferring canalization to a bistable patterning system in Drosophila.Gavin-Smyth J, Wang YC, Butler I, Ferguson EL. Curr Biol. 2013 Nov 18;23(22):2296-302. doi: 10.1016/j.cub.2013.09.055. Epub 2013 Oct 31.
